![David Molnar [Update:, PhD]](https://www.srcf.ucam.org/~dm516/wp-content/themes/twentyeleven/images/headers/chessboard.jpg)
[News: Someone linked this text from the Open Source Toolkit: Hardware article collection and channel of PLoS. Thanks.]
I built my first fluorescent microscope. A Leitz Labovert became a surprisingly decent fluorescent microscope for DIY after spending about £100 and 3D printing a few custom parts. It is a simplified design with only a barrier and an excitation filter, LED illumination and no dichroic mirror.
The quality is sufficient for observing fluorescent yeast and in general fairly faint Drosophila embryos in low-to-midrange magnification.
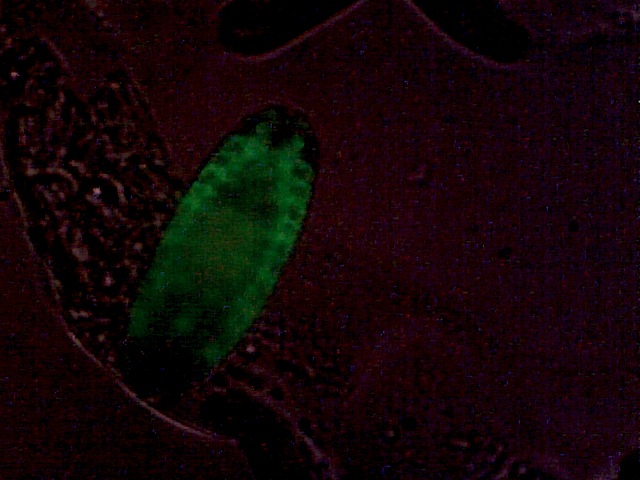
Drosophila embryonic tracheal system, GFP-marked. Weak additional bright-field illumination for embryo outline.
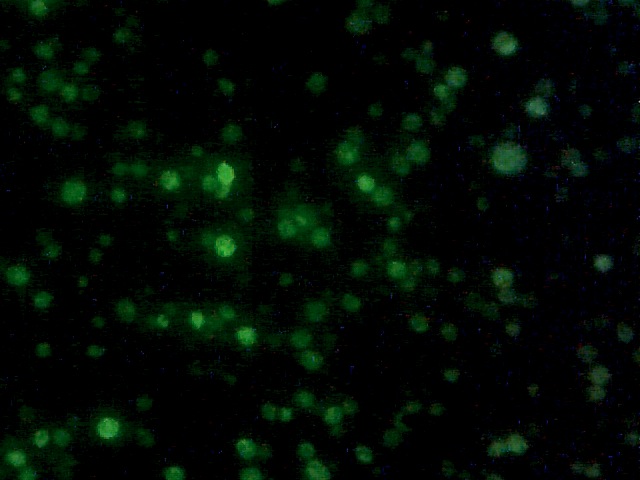
Yeast with nuclear GFP marker.
Fluorescence is short wavelength light in, longer wavelength light out. In principle a fluorescent microscope needs only two parts: 1) a light source that emits light at the excitation, but not at the emission wavelengths of the sample, and 2) a barrier filter, which lets only the emitted longer wavelength light through, so it doesn’t drown out in the excitation light. There are a few details though, and I will start with the barrier filter.
Barrier filter
The basis for the conversion was a Leitz Labovert. It is a fine microscope from the 80s and shares a lot of parts with its sister microscope, the Fluovert, making the conversion easier. According to the manuals (at Frank’s Hospital Workshop) both have the same vertical light duct in the back with a port where the dichroic mirror and the fluorescent light source latch in. This port is hidden in the Labovert behind the back cover plate. The duct is a good place for interference filters (more about them later), because these filters’s cut-on wavelength is dependent on the angle of incident light. Light travels as an entirely parallel beam of rays here, and thus the nominal cut-on wavelength of the filter applies everywhere. In a converging beam the cut-on wavelength would be shorter towards the rim than at the centre (more details here).

The Fluovert.



Instead of reconstructing the whole Fluovert module, I 3D-printed a slot and a drawer for the barrier filter. The drawer (AutoCAD, Makerbot) moves into the slot (AutoCAD, Makerbot) whenever fluorescence is needed, and out otherwise. Double-sided sticky tape keeps the snugly fitting slot in place.





For access to the drawer, the mechanical workshop of Chemistry Department kindly cut a hole into the back plate, where otherwise the port of the Fluovert would have been. To make it neat, I printed a little frame for the hole (AutoCAD, Makerbot). It is slightly asymmetric to make up for the tolerance of the hole.


The filter is a dielectric FEL0500 shortpass with cut-on at 500 nm from Thorlabs (£52.56). Thorlabs sells these filters in 50 nm steps, and luckily a 500nm cut-on is perfectly suitable for GFP and eGFP. (see figure below)
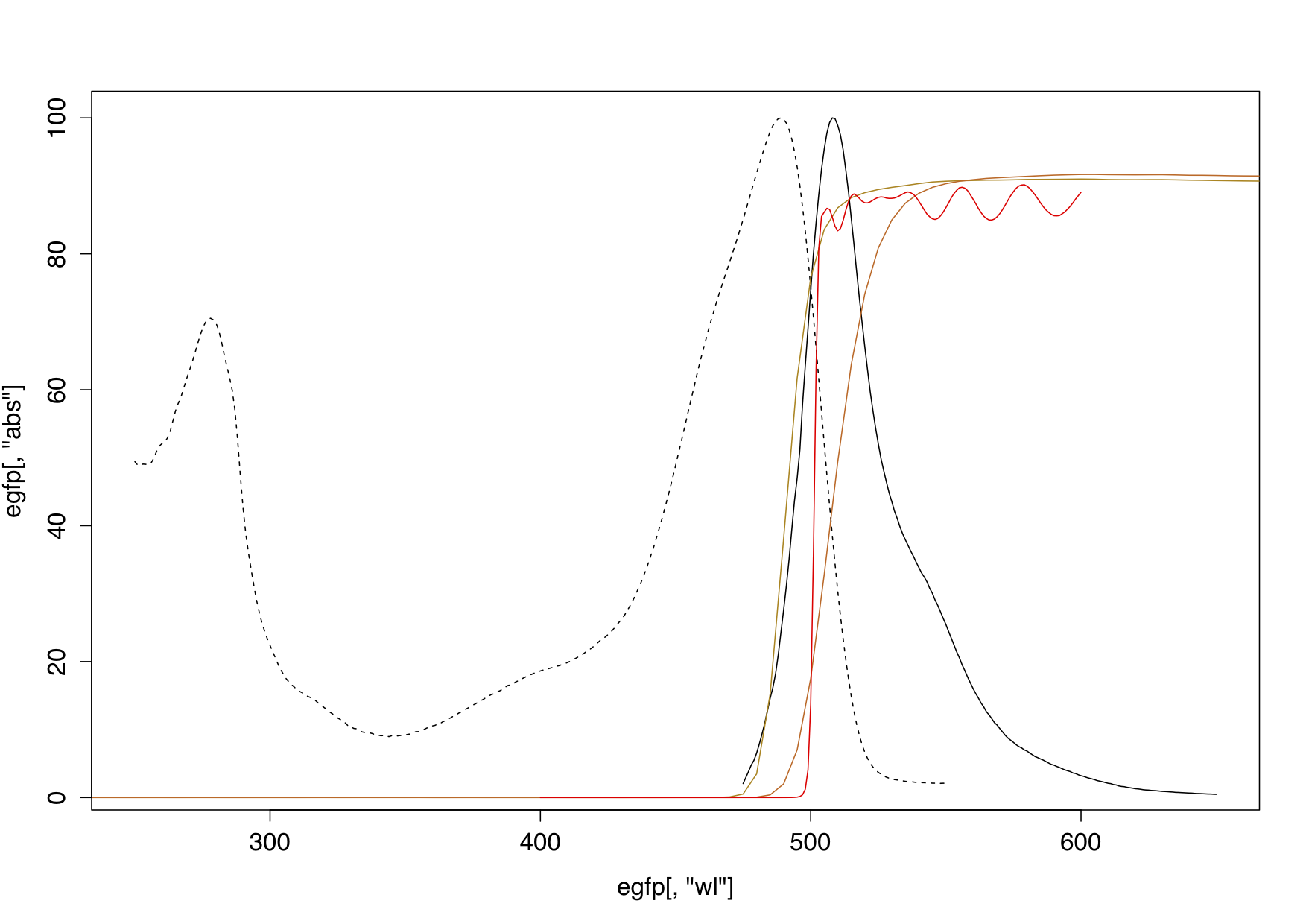
Different filter transmissions compared to eGFP absorption and emission. Red – FEL0500 transmission, gold – two different dyed glass filters for comparison, dashed black – eGFP absorption, solid black – eGFP emission. Transmission data from Thorlabs, GFP data from the Tsien Lab.
The FEL0500 has some good features for imaging, superior to any dyed glass filter. Most importantly it has a very steep cut-on edge (from nearly 0 to 90% transmission in ~8 nm). The contrast is a good ~1:10^6 between blocked and transmitting spectral regions. These two features are necessary for a bright image with much contrast. In addition the surfaces of the FEL0500 seem to be well-aligned, as the filter produced no discernible ghost images from internal reflection.
A filter contrast of ~1:10^6 may seem unnecessarily high to separate the tail end of the excitation spectrum from emission (fluorescence in a blue illuminated GFP solution is visible with naked eyes). An average microscopic sample however is not a very effective converter of light. The brightness of GFP is not a problem in itself, as GFP quantum yield is quite good at ~0.8 (10 blue excitation photons make on average 8 green fluorescence photons). The main problem is that there are not that many GFPs in the observed volume at all. The focused layer of a specimen is very thin (range of um) and in this layer actual fluorophore molecules (p-hydroxybenzylidene-imidazolidone in the centre of each GFP protein) are few and far between. Therefore only a few out of many excitation photons will hit a GFP, and produce an emission photon in the first place. Furthermore, emitted photons are undirected, and therefore only a fraction will reach the objective. In summary, the fluorescent image is very faint compared to the massive excitation light. With my samples the filter just began to give the meagre output an edge over input. Any reduction in excitation light that reaches the observers eye seemed welcome to increase sensitivity and make the faint fluorescent image stand out better. Improvement came, as discussed below, from the right illumination.
The resolution limit of the system became apparent with objectives more than 20-30x. No further details emerged at more magnification and the image became blurred instead. I presume this was for multiple reasons: the objectives, although made for fluorescence, are quite old, and the resolution provided by the illumination (discussed below) becomes limiting. Quite likely the resolution degradation by the not quite-for-imaging-filter became more apparent here, too.
Illumination
The illumination is a 1W Luxeon Royal Blue LED in a Lumidrives – LSP1-B-520 module (£6.09), with an added focusing lens and an excitation filter. The Luxeon Royal Blue covers the excitation ranges of both wtGFP (~380-440 nm) and eGFP (~450-500 nm) well. It requires a cooling body (OHMITE – SV-LED-325E, £3.20), which is held in place by Akasa AK-TT12-80 Thermal Adhesive (£2.92), basically a heat conducting double sided sticky tape. The LED is powered by a minimal circuit as suggested by the LED series parallel array wizard: a 4.5V power supply plug (£8.59) and a 3.3 Ohm resistor (Farnell) in line with the LED.
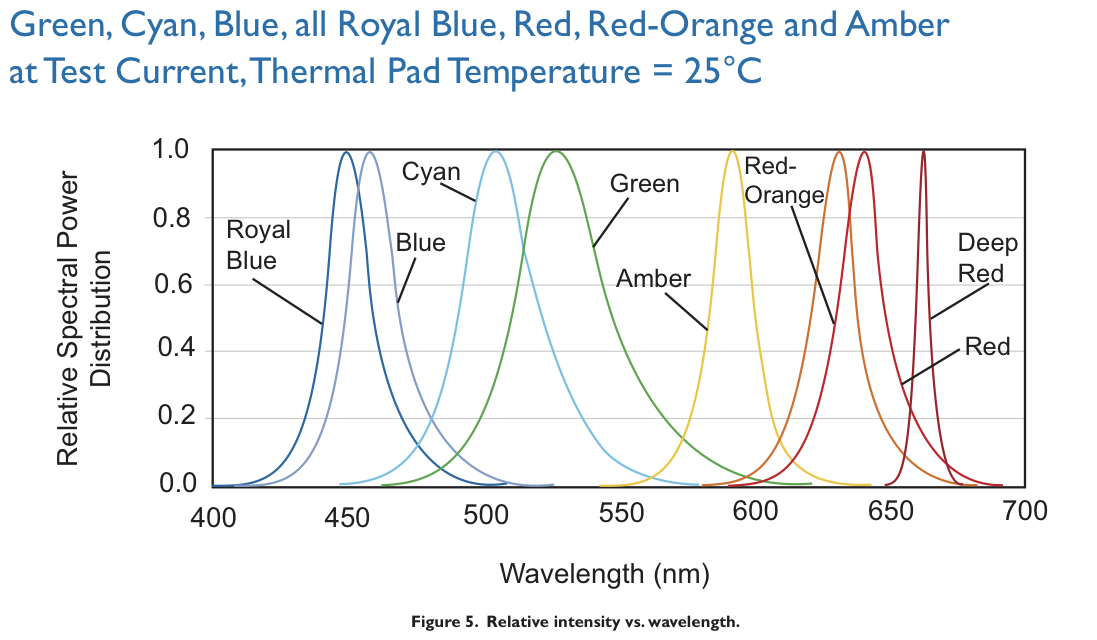
Emission spectra from the Luxeon Lumileds manual.
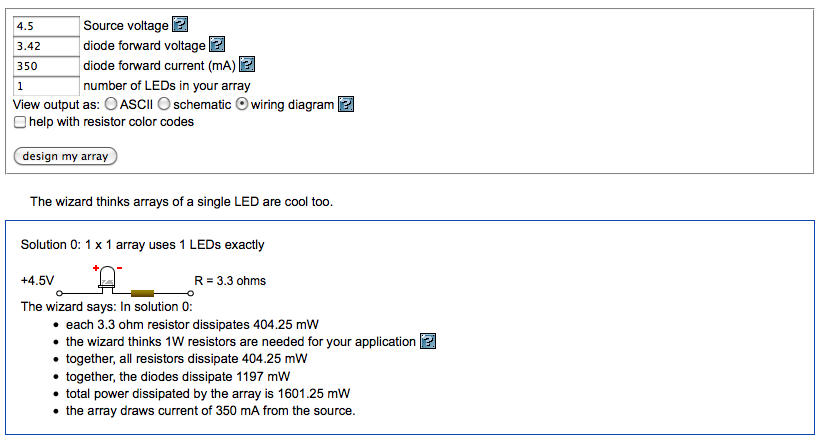
The linked Lumidrives module has a built in focusing lens, but the wrong one: not a 5° spot as the website suggests, but one that produces a slit of light. A replacement lens L2 OPTICS – OP-005 – LENS, SPOT, 20MM (£1.09) solved this problem, but the spot was still too large. A second focusing lens Comar Optics 62 VP 25 (£4.60) stuck to the first made a handsome focused spot on the microscope slide which was now reasonably bright.
The emission filter is an important component for image quality. When LED manufacturers publish emission curves like in the figure above, they are not strictly truthful. All blue and purple LEDs I have tested (Luxeon Royal Blue, Maplin Superbright Blue LED, a generic near UV from onecall) also emitted small amounts of green, yellow and red light all over the longer wavelength part of the spectrum. These may have been minor contributions, but unfortunately they were reflected back from the sample, went straight through the long-pass barrier filter, and were brighter than the fluorescence which I would have liked to observe. To block them out, I used a Comar Optics 495 IK 125 interference short-pass filter (£17) with a cutoff around 490 nm as emission filter. The filter was good enough here despite having only 1/10 of the contrast of the Thorlabs FEL0500, and a cutoff flank that is much less steep.

The contrast was further improved greatly by oblique illumination from below, to avoid getting illumination back into the objective. The usual way to illuminate is to use a dichroic mirror and send the excitation light through the objective. Out of the objective the excitation light goes up vertically through the specimen, and the unabsorbed bulk of the light then travels straight on to some sink. Whatever light comes back from the sample to the objective can only be fluorescence, reflection or scattering. The dichroic mirror gets rid of scattered and reflected short wave excitation light coming back. In my simplified setup however placing the LED sideways below the sample worked well, without a dichroic mirror. This position is better than oblique lighting from above, because excess illumination always went up, despite any possible refraction on the way, and any reflection on the glass slide went to the side, past the objective. The illumination contraption was held together and in place just by sticky tape and plastic foam right now, but it will eventually become a proper fixed setup.
Conclusions
Taken all together the quality has been in a good relation to the invested effort. If the Thorlabs filter had been a little thinner (by 1mm), or a little less wide (by 1mm) it would have fitted into either of the two drawers underneath the nosepiece turret, originally made for a polarising filter and a colour filter. This would have made the whole construction a lot simpler, and with access to a fitting filter, there is a chance to save some work here. Either way, it would probably be easy to add more filter drawers and LED illumination packs for other fluorescent proteins, such as RFP.
In comparison to another DIY fluorescence microscope design which can only detect strong fluorescence, the use of interference filters seems an improvement. The interference filters were only marginally more expensive than the dye excitation and barrier filters in that project, and well worth the expense for steeper flanks and better contrast.
The takeaway message I suppose is to use interference filters, even if they are cheap ones, spend a little more on the imaging filter if possible, and to illuminate from the side. Life technologies FLoid (sic) microscopes, as I discovered recently at a demo, are more sophisticated but use now the same arrangement with LED illumination from below and the side.
Thanks
I kindly got the Labovert from the neighbouring Watt Stem Cells laboratory, where it was on the way to be scrapped. There is a plaque on it saying it has been a gift in memory of Mary Marjorie Kemp. Sadly Mrs Kemp seems to have no google footprint, but I certainly would like to hear if someone knows more.
Many thanks go to Eddie Kinrade who helped sourcing all these parts and introduced me to 3D-printing, Martin Jahnke for discussion, Boris Adryan for materials, and the CUCC mailing list, which immediately becomes excited when someone asks about LEDs. Also thanks to the hands on microscopy course at LMU Munich.